Research Projects!
I. Cell Phenotypes and Chromosome Stability in vitro
Background
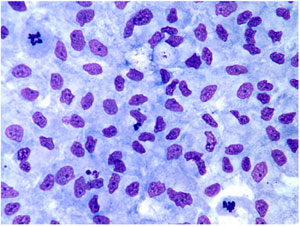
The classical ICR-2A established cell line from the amphibian, Rana pipiens, is used for the investigations. This line was developed from androgenetic haploid embryos by Freed and Mezger-Freed in 1970, and is available from the American Type Culture Collection as CCL 150. Current studies in my laboratory have shown that this cell line is a mixture of two different kinds of cells, one fibroblast-like in morphology, and the other, epithelial-like. Once separated from one another, the two grow as apparently pure cultures of each phenotype with no tendency to revert to the other or become a mixture again after continuous subculturing for over two years. The epithelial cells are small and grow as a monolayer of adherent cells that exhibit contact inhibition of division after reaching confluency, while the larger fibroblast cells form stellate clusters of cells that exhibit contact inhibition of movement and do not make confluent cultures (Fig. 1).
|
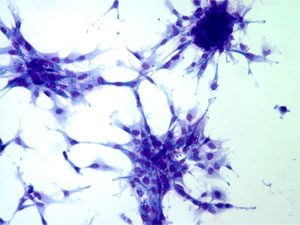 |
Fig.1. Left: Epithelial vs. Right: Fibroblastic cells.
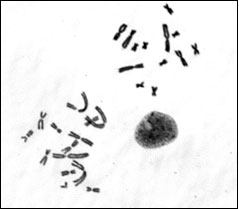
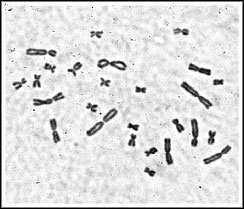
Chromosome contents. The epithelial line is a mixture of haploid, near-haploid, and cells of higher ploidy, while the fibroblast line is comprised of diploid or higher ploidy cells. The near-haploid cells are aneuploid for 14 instead of 13 chromosomes of the haploid karyotype, and the extra chromosome can be the nucleolar region-bearing chromosome 11 or a telocentric, small chromosome derived from a normal chromosome and now carried along as a chromosome fragment.
Fig.2. Left: Metaphase spread of the ICR-2A epithelial cell line showing cells with the haploid set of 13 chromosomes (upper right), and the aneuploid haploid set with 14 chromosomes (lower left) in the same preparation. Right: Metaphase spread of a diploid karyotype from the ICR-2A fibroblast line with 26 chromosomes.
Current studies:
1. Molecular cytology of the ICR 2A cell line—late replication banding, C-banding, NOR-banding. These studies will allow us to examine the types of intrachromosomal changes that accompany changes in ploidy as cells evolve in vitro.
2. Genetic manipulation of the ICR 2A cell line—expression of dominant selectable markers. These studies will allow us to determine the nature of multiple drug resistance observed in this established vertebrate cell line.
II. Lung abnormalities in Hermansky Pudlak Syndrome (HPS) mutant mice
Background:
HPS is a genetically heterogeneous disorder as more than one gene can cause this syndrome. The disease is characterized by hypopigmentation, prolonged bleeding time, and fibrotic lung disease in humans. Pulmonary fibrosis is the most critical clinical problem for HPS patients and often leads to premature death in mid-life. Some 15 different autosomal mouse mutants result in HPS-like traits, and 8 of these are known homologs to genes causing this disorder in humans. These include pale ear (HPS1), pearl (HPS2), along with six other pigment mutant genes. The pearl gene (HPS2 or AP3B1) encodes the beta3A subunit of the AP-3 adaptor protein complex, which regulates vesicle trafficking. The products of the other seven established HPS homologs are novel, including the pale gene, and presumed to play some role in vesicle trafficking.
HPS and lung fibrosis: Recently, an HPS mutant mouse was produced by appropriate breeding which is doubly homozygous for the pearl (Ap3b1pe) and pale ear (ep) genes in the BL6 genetic background: C57BL/6-Hps1ep Ap3b1pe-J. This double mutant mouse develops characteristics that are strikingly similar to those seen in HPS patients. These include an enlargement of lamellar bodies of alveolar type II cells, the secretory cells of the lung that are responsible for the production and regulation of surfactant materials. The enlargement of these bodies, which is also seen in the beige (CHS) mouse, is due to a genetic block to their secretion, a characteristic common among most HPS mice, and of the CHS mouse as well.
Chediak-Higashi Sydrome (CHS): CHS is apparently a monotypic or genetically homogeneous disorder, as a single gene is involved – the Lystbg, or beige gene, of the mouse. Mutants in both humans and the mouse have a defect in the same gene that encodes the beige protein and exhibit enlarged lysosomes due to the genetic block in lysosomal trafficking. Hence, the Lyst designation for this gene in the mouse, which is called CHS1 in humans. CHS patients have severe immune deficiencies that usually result in early death due to pneumonia in infancy and lymphoreticular malignancy in puberty or early adulthood.
Lung study: The initial investigations of the C57BL/6-Hps1ep Ap3b1pe-J HPS mouse were made in collaboration with Dr. Richard T. Swank of the Roswell Park Cancer Institute in Buffalo, NY, and showed that it exhibited lung abnormalities (Lyerla et al., 2003). This was an important first step in developing an authentic HPS mouse model for pulmonary fibrosis in humans, and we are concentrating our efforts in studying this model further, as well as testing for lung fibrosis in the beige CHS mouse. The beige mouse shares the distinct genetic block to secretion in the lung that is the hallmark of the HPS lung phenotype in both humans and the mouse model, and may be a factor in the development of the ultimately fatal pulmonary fibrosis seen in HPS patients.
Our recent investigations have shown that the double mutant HPS mouse is more susceptible to bleomycin-induced lung fibrosis than the CHS mouse, and that HPS mice older than one year of age can develop pulmonary fibrosis, whereas CHS and control C57BL/6J mice do not develop this disease upon aging. The production and secretion of TGF beta-1 by highly activated alveolar macrophages was considered of primary significance to the development of pulmonary fibrosis in these animals.
Current studies:
1. The nature of activation of the HPS alveolar macrophages.
2. The role of ATII cells in the activation of HPS alveolar macrophages.